
Genomic surveillance has revealed that malaria resistance to two first-line antimalarial drugs has spread rapidly from Cambodia to neighbouring countries in Southeast Asia. Researchers from the Wellcome Sanger Institute, University of Oxford and Mahidol University, Bangkok, discovered that descendants of one multi-drug resistant malaria strain are replacing the local parasite populations in Vietnam, Laos and northeastern Thailand. They also found the resistant strain has picked up additional new genetic changes, which may be enhancing resistance even further.
The study, published in The Lancet Infectious Diseases today (22nd July), reveals the importance of ongoing genomic surveillance to inform public health malaria control strategies. Global efforts to eliminate malaria could be threatened by any delays in detecting and acting on the spread of resistance.
Malaria is caused by Plasmodium parasites which are spread through mosquito bites. The World Health Organization estimates that nearly 220 million people were infected in 2017, causing at least 400,000 deaths*, with children under the age of five in sub-Saharan Africa at most risk. Malaria can be treated when caught early enough, but the parasite is becoming resistant to antimalarial drugs in many areas, especially Southeast Asia, putting elimination efforts at risk.
Over the last decade, the first-line treatment for malaria in many areas of Asia has been a combination of two powerful antimalarial drugs - dihydroartemisinin and piperaquine - otherwise known as DHA-PPQ. However, a previous study identified a strain of malaria that had become resistant to this treatment. Researchers found that this resistant strain, named KEL1/PLA1 because of its combination of genetic mutations that cause resistance, had spread across Cambodia under the radar between 2007 and 2013.
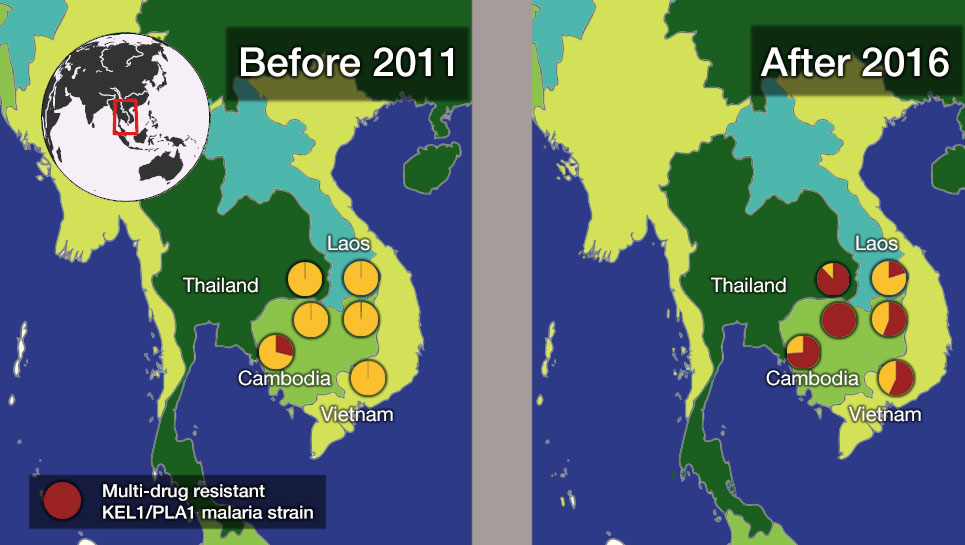
This highlighted an urgent need to follow up and determine how far this resistance had then spread and if it had evolved further, and eventually understand which drugs would work against current malaria parasites in Southeast Asia.
In the most up-to-date and comprehensive whole genome study of malaria parasites in Southeast Asia, the team sequenced and analysed the DNA of 1,673 Plasmodium falciparum parasites, taken from the blood of malaria patients between 2008 and 2018. Their analysis, focusing on the KEL1 and PLA1 gene variants, revealed that the situation had got much worse after 2013. The multidrug resistant KEL1/PLA1 parasites had spread internationally, in some regions making up more than 80 per cent of the parasites analysed.
Dr Roberto Amato, joint first author from the Wellcome Sanger Institute, said: “We discovered that the multi-drug resistant KEL1/PLA1 malaria strain had spread aggressively, replacing local malaria parasites, and had become the dominant strain in Vietnam, Laos and northeastern Thailand. Our large-scale genomic approach demonstrates how surveillance can provide crucial information to malaria control programmes, supporting them in evaluating available treatment options.”
The spread is likely to have occurred because resistant parasites had an evolutionary advantage, as DHA-PPQ was the first-line treatment in most of these areas. This killed other malaria strains but was less effective against KEL1/PLA1malaria.
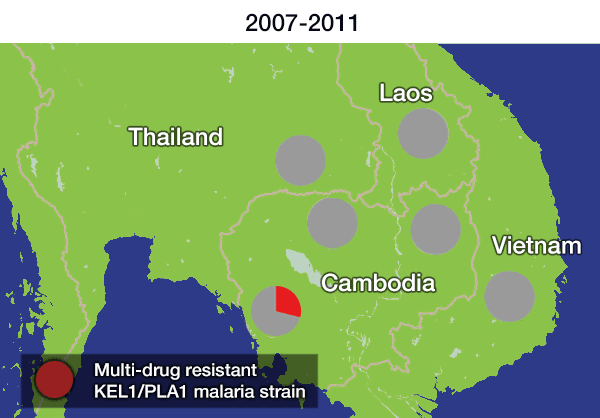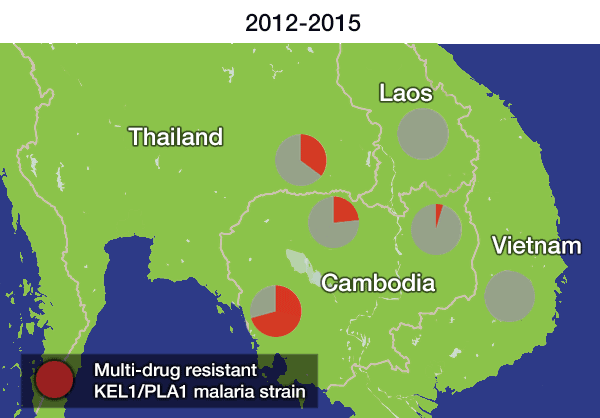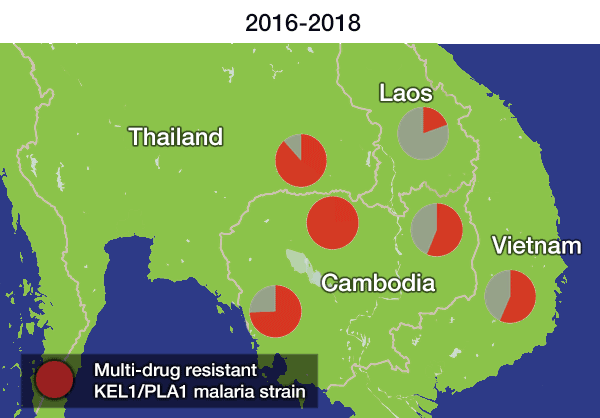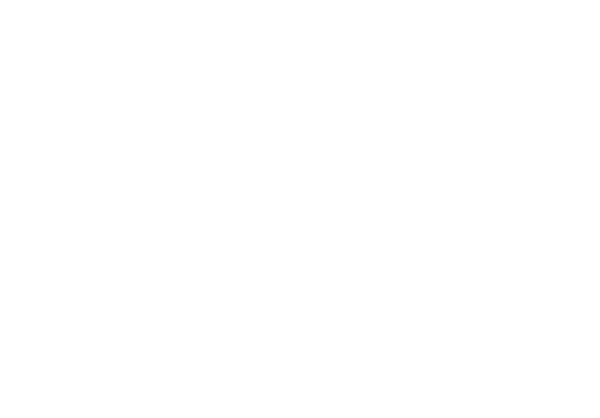
The researchers discovered that not only had this resistant strain spread geographically, but it had evolved and picked up new mutations in the chloroquine resistance transporter gene (crt). These parasites had then rapidly spread across borders. A related paper** on clinical outcomes published today in The Lancet Infectious Diseases revealed that these crt mutations were associated with complete treatment failure of DHA-PPQ. This supported the finding that the resistance had not only spread, but worsened as the parasite evolved under further drug pressure.
Professor Olivo Miotto, a senior author from the Wellcome Sanger Institute, University of Oxford and Mahidol University, Bangkok, said: “The speed at which these resistant malaria parasites have spread in Southeast Asia is very worrying. Other drugs may be effective at the moment but the situation is extremely fragile and this study highlights that urgent action is needed to eliminate the parasites from the Greater Mekong Subregion, to prevent them spreading and evolving further.”
Dr Michael Chew, Infection and Immunobiology Portfolio Manager at Wellcome said: “This study clearly shows the rapid spread of multi-drug resistant malaria across Southeast Asia. Affecting millions globally, malaria is a devastating disease, especially when access to effective treatment is unavailable.
“The spread of drug-resistant strains as demonstrated by this important genetic surveillance, building upon previous research, provides a warning signal. It serves as a reminder that we must not show complacency with the response to malaria. It also shows we have the tools to effectively track drug resistance across borders, which can be used to inform co-ordinated elimination and control efforts.”
Professor Dominic Kwiatkowski, a senior author on the paper from the Wellcome Sanger Institute and the University of Oxford, said: “Our study provides a clear picture of how malaria that is resistant to the first-line treatment is spreading, and demonstrates the importance of using genetics to detect patterns of resistance in each area. Active genomic surveillance is now vital to inform national malaria control programmes, to help reduce the risk of a major global outbreak.”
Notes to Editors
Publication:
William Hamilton and Roberto Amato et al. (2019) Evolution and expansion of multidrug resistant malaria in Southeast Asia: a genomic epidemiology study. The Lancet Infectious Diseases. DOI 10.1016/S1473-3099(19)30392-5
Footnotes:
*Figures from the 2018 World Health Organisation report https://www.who.int/malaria/publications/world-malaria-report-2018/en/
**Rob van der Pluijm et al. (2019) Determinants of dihydroartemisinin-piperaquine treatment failure in falciparum malaria in Cambodia, Thailand and Vietnam: a prospective clinical, pharmacological and genetic study. The Lancet Infectious Diseases. DOI 10.1016/S1473-3099(19)30391-3
Funding:
This work was supported by Wellcome (098051, 206194, 203141, 090770, 204911, 106698/B/14/Z), Bill & Melinda Gates Foundation (OPP1040463, OPP11188166), Medical Research Council (G0600718), UK Department for International Development (201900, M006212), and other funders. Please see the paper for full funding details.
Contact
Dr Samantha Wynne,
Wellcome Sanger Institute
Hinxton, Cambridge, CB10 1SA, UK
Tel +44 (0)1223 492368
Email press.office@sanger.ac.uk


