

In order to beat malaria, we must understand it. To that end, the latest MalariaGEN P. Falciparum dataset — called Pf7 — was published this week in Wellcome Open Research.
This huge data resource is a collaboration of more than 150 partners from the MalariaGEN community, and includes samples from 33 different countries, collected between 1984 and 2018. The paper provides clean, analysis-ready data, along with a series of preliminary investigations of drug resistance markers, the regions targeted by the new WHO-approved RTS,S vaccine, and hrp2 and hrp3 deletions.
Along with free-to-download raw data files, the MalariaGEN team have produced a python package that uses cloud computing via Google Cloud to enable analysis without downloading the full dataset. All one needs to access and start using the data is a laptop and an internet connection.
Open the Pf7 app to view summary information about contributing studies, countries, and resistance profiles.
“We are very proud of this release, and the community that has come together to build it” says Principal Data Scientist Dr. Richard Pearson, who is the corresponding author for the paper. “I see Pf7 as a springboard, from which many important discoveries can be made. Very early analyses have already turned up interesting findings that policymakers may wish to take into consideration.”
Highlights

- Pf7 is the world’s largest data resource for P. falciparum genomic information
- The number of samples has nearly tripled since the last release in 2020, from 7,112 to 20,864
- The dataset includes samples from 82 partner studies in 33 countries in Africa, Asia, South America, and Oceania between 1984 and 2018.
- New methods were developed to include genomes extracted from dried blood spots (DBS).
- All data is freely available to download at malariagen.net
Malaria is a complicated disease caused by a microscopic parasite transmitted between humans by mosquitoes. It kills hundreds of thousands of people every year, mostly young children and pregnant women in sub-saharan Africa. The toll is so great that if malaria deaths were spread evenly over the course of the year, one child in Africa would succumb every minute.
While there are several species of human malaria parasite, by far the most prevalent and deadly is Plasmodium falciparum. In 2021, it caused approximately 98% of the 247 million cases worldwide. Wherever it appears, people use a variety of drugs, insecticides, and other strategies to stop its spread. These control measures exert different evolutionary pressures, which will show up in the genome. By keeping track of large-scale genetic variation, we can spot when the parasite begins to evade our drugs, vaccines, or other measures.
The Malaria Genomic Epidemiology Network (MalariaGEN) was established in 2005 to serve as a central clearinghouse for malaria genomic data. One of the first large datasets, published in Nature in 2012, contained whole genomes of 227 P. Falciparum samples.
Technological leaps
Since then, both the network and the datasets have grown. The latest iteration now includes whole genome sequences from 20,864 samples, including more than 12,000 from dried blood spots. The technology to extract whole sequences from portable and easy-to-collect dried blood spots was developed at the Sanger Institute by a team co-led by Cristina Ariani, who is now the MalariaGEN Malaria Parasite Surveillance Lead.
“We are thrilled to publicly release this dataset, along with some fascinating analysis” says Dr. Ariani. “We hope that the scientific community can use this information to identify new ways to fight malaria. We’re also keen to increase the pace of data releases, so you’ll be hearing more from us sooner rather than later.”
Part of the reason for the longer than normal delay between Pf6 and Pf7 was to do with making sure the data was clean and unbiased. Because the parasite DNA on bloodspots had to undergo more processing and amplification, there was a worry that the DBS data wouldn’t be as high-quality as the more traditionally-extracted venous blood DNA. With some clever computing, this fear proved unfounded.
“If there were going to be problems [with the data from bloodspots], we would have seen them on the whole genome analysis. We would have been able to tell the difference between genomes extracted from bloodspots and those from venous blood draws,” says Dr. Pearson, who oversaw rigorous tests to ensure that the data from bloodspots weren’t tainting the database. “But the samples appear completely mixed together. We couldn’t identify an effect.”
Preliminary analysis
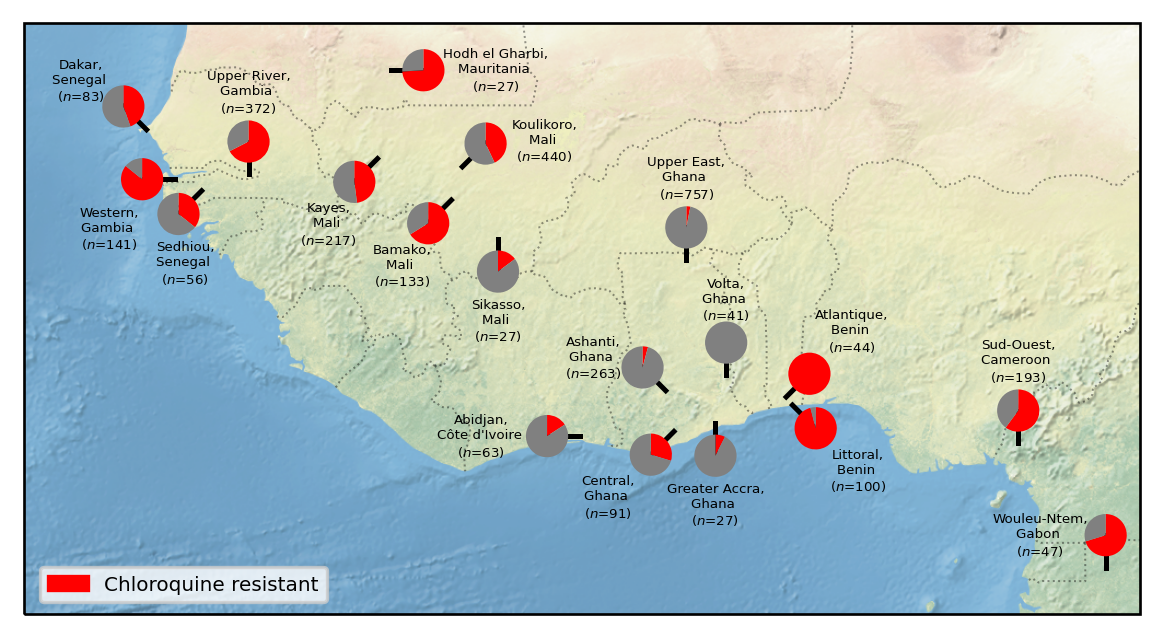
- Drug resistance marker maps are included, and show surprising heterogeneity, even between nearby countries (e.g. parasites in Ghana are nearly all chloroquine-sensitive, while those in Benin are nearly all resistant.)
- Preliminary analysis shows that mutations so far discovered do not significantly alter proteins targeted by the RTS,S vaccine, although the gene regions do show high variability.
- Data shows where on the chromosomes hrp2 and hrp3 deletions occur. There is a tendency for them tend to happen at the very end of chromosomes
Drug resistance
A major advantage of large-scale genomic sequencing is the ability to track changes in drug resistance profiles. This includes both how parasites vary across regions, as well as an increasing number of time series: how parasites in the same region vary over time.
The Pf7 release includes many drug resistance markers that show significant variation over both time and space. Even in sample locations that are physically quite close, sometimes parasites are genetically distinct and are susceptible to different drugs. This highlights the need for integrating genomic surveillance into drug policy decisions.
Vaccine-targeted regions
With the WHO recommending the RTS,S vaccine in 2021 and the R21 vaccine showing promising results from field trials, a new class of anti-malaria intervention has entered the picture. These vaccines will undoubtedly affect parasite genetics. In Pf7, the MalariaGEN team demonstrated a quick and inexpensive way to check genetic variation in the regions targeted by vaccines.
Analysis of the CSP region of the genome, the proteins for which are targeted by both RTS,S and R21, showed significant variation between samples. This analysis could easily be used for future vaccine development to check how well the candidates match parasites circulating in the field.
Rapid tests
The vast majority of commonly-used rapid diagnostic tests (RDTs) for malaria detect the presence of the proteins HRP2 or HRP3, which are usually produced in abundance by P. falciparum parasites. Some parasites, however, are missing these proteins. Exactly where and how the genes that code for HRP2 and HRP3 disappear is an active area of investigation.
In analyses published with the Pf7 dataset, the MalariaGEN team have confirmed that the deletions are occurring at the ends of the chromosome, right before the telomere. Further, they show that there are several different places where the breakages are happening. By tracing which breakage is occurring, epidemiologists can now infer genetic history. In other words, they can determine whether a strain of RDT-evading parasites was imported into a country or whether it evolved there independently.
These three preliminary analyses are proofs of principle, showing the kinds of public health impact that large-scale genomic resources like Pf7 can have. They are meant as instructive examples rather than exhaustive investigations. The power of community resources like Pf7 and MalariaGEN is in understanding the full genetic picture of malaria. This allows more tailored control measures and lets public health officials spot threats earlier.
Read the paper introducing Pf7 in Wellcome Open Research: https://wellcomeopenresearch.org/articles/8-22
To download the full analysis-ready data, visit the data page.

