
Despite huge efforts to treat and eradicate the disease, in 2015, 214 million people were infected with malaria. 438,000 died. More than 292,000 of those deaths were African children aged under five. Treatment is complicated by the fact the malaria parasite develops resistance to antimalarial drugs.
Oxford researchers are looking for new ways to target malaria, including vaccines and drugs, but the answer may lie in genes – our genes, the parasite’s genes and even those of the mosquito that carries it. A team based in Oxford and at the Wellcome Trust Sanger Institute, just outside Cambridge, are working together with scientists and clinicians in more than 40 malaria-endemic countries to integrate epidemiology with genome science.
This is the Malaria Genomic Epidemiology Network (MalariaGEN), a global researcher community aiming to understand the genetic diversity in the human host, the parasite and the mosquito vector.
In the second of two interviews about research from MalariaGEN, we look at a study published in eLife, which looks at the genetics underlying the developing resistance to artemisinin.
Central to this is something called kelch13, so when I spoke to Dr Roberto Amato, I started by asking what that is…
Roberto Amato: kelch13 is one of the ‘nicknames’ of a gene officially known as 'PF3D7_1343700' in the malaria-causing parasite Plasmodium falciparum. The gene sits more or less in the middle of chromosome 13 and contains a particular sequence which is found in many species including humans, the 'kelch motif' – hence the name 'kelch13'.
This gene is highly conserved in the Plasmodium genus; its DNA sequence has remained nearly unchanged for more than 50 million years.
The gene has been the focus of much attention since December 2013, when research led by the Institut Pasteur first associated mutations in this gene, and specifically within the kelch domain, with resistance to artemisinin, the current frontline treatment for uncomplicated malaria.
Since then, several studies have confirmed that this gene plays a crucial role in artemisinin resistance in clinical samples while others have focused on figuring out how the gene works. Much is still unknown but one thing is clear: this is an unusual beast, and a very complicated gene to deal with! In fact, what has gradually emerged is that several mutations to the gene all cause resistance to the drug.
We know that P. falciparum mutates rapidly, so the question becomes: Why didn’t resistance emerge earlier – and everywhere? It's possible that there is a more complicated story that we have yet to understand.
OSB: This does sound complex. How do you start to make sense of mutations in kelch13?
We decided to study not only this one gene, but to look across the whole parasite genome.
We're fortunate enough to work with researchers in dozens of malaria-endemic countries through the MalariaGEN P. falciparum Community Project. Together, this research network has generated a large — possibly the world's largest — collection of whole genome sequences of P. falciparum parasites. This resource allowed us to scan 3,411 parasite genomes from samples collected in 23 countries.
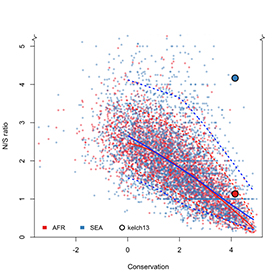
This large sample size meant that we could 'calibrate' the mutations that we could detect in kelch13 against other genes with similar levels of conservation. This is important because there are few observable mutations in highly-conserved genes, so we need large sample sizes to find enough genetic changes to achieve adequate statistical power.
In particular, we want to compare the mutations present in Southeast Asia, where artemisinin resistance has been reported in several countries, with Africa, where malaria transmission rates are highest but, as far as we know, there aren't yet signs of clinical resistance.
We found three interesting differences between the genetic variations observed in parasites from these two regions:
Firstly, we showed that some of the key mutations associated with artemisinin resistance in Southeast Asia, most notably C580Y, are already present in Africa.
Previous studies have shown that mutations in kelch13, and in particular in the kelch domain, were present in Africa. However, these were different mutations than those found to be associated with artemisinin resistance in Southeast Asia. This gave rise to the suspicion that 'not all mutations are created equal'. While this might well be true, our results challenge this hypothesis — and highlight once again the complexity of the intricate (and intriguing) story of artemisinin resistance.
Secondly, these key mutations associated with artemisinin resistance in Southeast Asia are less common in African parasites.
Using our large data set, we were able to determine what constitutes a 'physiological' level of variation that is consistent with our expectations – essentially, how common we'd expect these mutations to be in the absence of a selective pressure. This is what we observe in African samples, but the key kelch13 mutations are far more common in Southeast Asia. This is an intriguing difference which I'll come back to in a moment.
Thirdly, in Southeast Asia, we observed more non-synonymous mutations than what we expected, but not in Africa.
OSB: What might be the reasons for these differences?
RA: Non-synonymous mutations are interesting because they have the potential to produce drastic changes to the protein and the way it works (in contrast with synonymous mutations that have no effect on the resulting protein). For this reason, non-synonymous mutations are typically less common in highly-conserved regions like kelch13. Their unusually high presence in Southeast Asia suggests that there is a reason, possibly a selective pressure, driving this pattern of genetic variation.
We think that 'something' allows kelch13 mutations to occur more often in Southeast Asia but not in Africa. We don't know what that something is, but we have some ideas – and these hypotheses are not necessarily mutually exclusive.
For example, drug pressure in Africa is not as sustained as in Southeast Asia and it might not be sufficient to counterbalance the likely fitness cost of kelch13 mutations – don't forget that if kelch13 has remained nearly unchanged for so long, there must be a reason! This lack of selective pressure could be because access to artemisinin in Africa is more recent and still limited. But it may also have to do with the acquired immunity that adults often develop in regions where malaria is common. In high-transmission areas, such as Africa, people may become infected without realising it. The result is a large reservoir of parasites that are not exposed to antimalarial drugs because individuals without symptoms do not seek treatment.
Another possibility is that other changes in the parasite genome have emerged in Southeast Asia, but not yet in Africa, to compensate potential deleterious effects of kelch13 mutations. Some evidence for this is provided by our earlier paper in Nature Genetics.
OSB: What does this mean for malaria control?
RA: Artemisinin resistance has been reported in several countries in Southeast Asia, which is leading to increased interventions in many of the affected areas. If artemisinin resistance were to become established in Africa (either through spread or independent emergence), where malaria transmission is highest, it could trigger a public health crisis.
Our study is an important reminder that malaria control interventions, like the administration of drug treatments, create a changing 'environment' – from the parasites' point of view, what we call drug resistance is simply adaptation to a new environment. Malaria parasites have proved tremendously good at that: resistance has emerged to every antimalarial drug used to date! Through this lens, drug administration can be seen as a large-scale evolutionary experiment.
Monitoring molecular markers of drug resistance in malaria parasites can help to track the emergence and spread of drug resistance. Some molecular markers are straightforward to monitor. For example, chloroquine resistance can be rapidly identified by typing a single base change in the crt gene in P. falciparum.
What we're seeing is that the signals are much more complex in the case of artemisinin resistance. Our findings suggest that the pattern of genetic diversity in kelch13 is not straightforward, and to use it as a molecular marker means 'contextualising' observed mutations. That in itself is an important message for those considering how molecular studies can help inform malaria control and respond to the challenge of artemisinin resistance.
OSB: What does this research tell us about the future for antimalarial resistance? And the future for the fight against malaria?
RA: If we buy into the idea that drug interventions are evolutionary experiments (at least from the perspective of the parasite) then we should do our best to try to evaluate, and possibly anticipate, the impact of such interventions.
As drug resistance can be extremely complex and dynamic, we need to use every arrow in our quiver to counter it. Genomics is one these arrows. And, not insignificantly, it is also one of the few that allow us to directly observe how the parasite is responding without many of the confounders stated before (e.g. acquired immunity, differences in transmission settings, etc).
If we are to integrate genomic surveillance into large-scale public health interventions, this would help to increase the resolution of our picture of genetic variation in space and time – and provide a valuable feedback loop of information for malaria control.
The paper, Genomic epidemiology of artemisinin resistant malaria, is published online in the journal eLIFE (DOI: 10.7554/eLife.08714).

