
Despite huge efforts to treat and eradicate the disease, in 2015, more than 200 million people were infected with malaria. Nearly half a million people died. The current frontline treatment for malaria is a drug called artemisinin but treatment is complicated by the fact that the malaria-causing parasite Plasmodium falciparum is exceptionally good at developing resistance to antimalarial drugs.
Artemisinin resistance has been reported in several countries in Southeast Asia. However in Africa, where malaria transmission rates are highest, there are no obvious signs of clinical resistance. If artemisinin resistance were to become established in Africa, it could trigger a public health crisis.
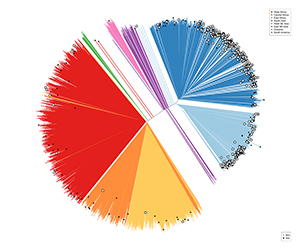
Two years ago, several mutations in a gene called kelch13 were implicated in artemisinin resistance. Although it might sound a bit counterintuitive, we realised that to better understand variations in this gene we had to look across the whole genome of a lot of parasites. Working with researchers in malaria-endemic countries through the MalariaGEN P. falciparum Community Project, we scanned more than 3,000 parasite genomes from samples collected in 23 countries, which were all whole-genome sequenced here at the Wellcome Trust Sanger Institute.
We know that, genetically speaking, there is a proliferation of new mutations occurring de novo in the Plasmodium falciparum genome all the time, regardless of where a parasite is from (Claessens, Hamilton et al, 2014). This creates a reservoir of underlying mutations which can be used to ‘calibrate’ the analyses and judge, for example, whether a mutation appears at a higher frequency at a given location.
Our aim was first of all to understand if kelch13 mutations observed in Southeast Asia were also present in Africa and, if so, what was their origin. Eventually, we wanted to compare those mutations across continents. Across the genome, we see a wider variety of mutations in Africa, while more mutations are pushed to higher frequency in Southeast Asia.
Our analysis, published this week in eLife, highlighted interesting differences between the genetic variations observed in parasites from these two regions of the world.
Firstly, we showed that some important mutations in kelch13 that are associated with artemisinin resistance in Southeast Asia, most notably C580Y, are indeed present in Africa. Leveraging on the fact that we were able to analyse the entire genome, we showed that these mutations are indigenous to Africa. That is to say that they emerged there. This challenges the notion that the apparent absence of artemesinin resistance in Africa is simply because the associated mutations aren’t present.
In line with what we observed at the genomic level, we also noticed that despite the variety of kelch13 mutations occurring in Africa, they tend to be rare. On the other hand, kelch13 mutations are pushed to higher frequency in Southeast Asia.
Finally, we observed more non-synonymous mutations than we expected in Southeast Asia, but not in Africa. Non-synonymous mutations [https://en.wikipedia.org/wiki/Nonsynonymous_substitution] change the resulting protein and are typically less common in genes that are highly conserved, such as kelch13.
Taken together, this seems to suggest that ‘something’ allows kelch13 mutations to emerge and reach high frequency in Southeast Asia but not in Africa. It could be, for example, the type and level of drug use, the malaria transmission setting, or the different genetic background of the parasite. It is almost certainly the product of multiple factors.
Bringing together parasite genetic data in a systematic and prospective way over the coming years will provide a framework to not only monitor what’s going on, but also to understand why and how mutations get selected.
Our study is also an important reminder that malaria control interventions, like the administration of drug treatments, create a changing ‘environment’ for the parasites. Seen from the parasite’s viewpoint, what we call drug resistance is simply adaptation to a new environment. In a way, drug administration can be seen as a large-scale evolutionary experiment and we should do our best to try to evaluate, and possibly anticipate, the impact of such interventions. Integrating genomic surveillance into large-scale public health interventions would help to provide a valuable feedback loop of information for malaria control.
Roberto Amato is a Staff Scientist in Dominic Kwiatkowski’s group and is involved in the analysis of natural genetic variation in the Plasmodium parasites that cause malaria. His primary focus is at the intersection of theoretical and empirical approaches aimed at characterising and understanding the evolution of the malaria parasite and its clinical consequences such as the emergence of drug resistance. Based in the Wellcome Trust Sanger Institute’s Malaria Programme, Roberto works closely with the colleagues at the Wellcome Trust Centre for Human Genetics at University of Oxford and supports several global collaborations including the MalariaGEN P. falciparum Community Project and Pf3k.
References:
- MalariaGEN Plasmodium falciparum Community Project (2016) Genomic epidemiology of artemisinin resistant malaria. eLife. DOI: 10.7554/eLife.08714
- Claessens, Hamilton et al. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of Var genes during mitosis. PLoS Genet. 2014 Dec 18;10(12):e1004812. DOI: 10.1371/journal.pgen.1004812
